
 Open Access
Open Access Abstract
Background: Rett syndrome is a rare disease but affecting severe brain dysfunction. The rate of the syndrome is estimated to be 1/10,000–1/15,000 female births. This disorder is knowledge caused by de novo mutations in the MECP2 gene. More than 80% of patients with Rett syndrome have a point mutations in MECP2. The study correlation genotype-phenotype open to therapeutic opportunities for patients. In Vietnam, to validate the diagnosis, the doctors are mainly clinical diagnostic standards, not include genetics testing. Therefore, optimize the producer to determine MECP2 variations is necessary that provides evidence for clinical diagnosis, supporting the earlystage diagnosis of pediatric patients.
Methods: Standardization of the MECP2 gene testing sequence used the healthy control blood samples includes PCR optimization and Sanger sequencing. Read raw sequence data by CLC workbench software. Analysis variations; evaluate the pathogenic of the variants with ACMG standard. After that, the process was applied to 4 pediatric patients with Rett syndrome treated at Children's Hospital II from 01/2019 to 12/2019.
Result: We successfully optimized PCR-Sanger sequencing for the testing MECP2 variants gene, especially in exon 1 had high GC-percent (> 65%). The quality sequencing result was quite reliable. We found two pathogenic mutations (p.R294X, p.K29X); and a novel mutation (p.K29X).
Conclusion: The process of the test gene MECP2 by PCR technique-Sanger sequencing was optimized completely, and add a novel mutation to the MECP2 variation database.
GIỚI THIỆU
Hội chứng Rett (RTT, OMIM # 312750) là một bệnh lý ở não gây ra các rối loạn về phát triển thể chất và thần kinh nghiêm trọng được miêu tả lần đầu vào năm 1966 bởi bác sĩ Rett 1 . Bệnh nhân được sinh ra trong thai kỳ khỏe mạnh, tiền căn gia đình bình thường, trẻ phát triển bình thường sau sinh. Sau đó, khoảng 6-18 tháng tuổi trở đi trẻ bắt đầu xuất hiện các triệu chứng chậm phát triển, hạn chế giao tiếp bằng cử chỉ và ngôn ngữ, đặc biệt bằng mắt, giảm khả năng trí tuệ và có biểu hiện tự kỷ. Ngôn ngữ của trẻ bị mất một phần hoặc toàn bộ. Trẻ mắc hội chứng Rett giảm khả năng vận động, mất dần khả năng sử dụng bàn tay có chủ đích, có những cử động tự động như xoắn vặn bàn tay và các ngón tay. Ở giai đoạn muộn sau 3 tuổi, các rối loạn vận động, loạn trương lực cơ tiến triển nặng hơn gây bất thường dáng đi và cong vẹo cột sống, giảm sức cơ và tư thế bất thường. Các bệnh nhân thiếu đi một hoặc một số đặc điểm lâm sàng chính của hội chứng Rett điển hình 2 , 3 được gọi là mắc hội chứng Rett không điển hình 2 . Ngoài ra, trẻ còn có các biểu hiện bệnh khác như co giật, khởi phát động kinh sau 3 tuổi, đầu nhỏ, tăng trưởng chậm, bất thường kiểu thở, rối loạn nhịp mạch, khó nuốt, rối loạn giấc ngủ và hành vi,… 2 . Việc chẩn đoán lâm sàng hội chứng Rett thường rơi vào giai đoạn muộn và dễ nhầm với các rối loạn phổ tự kỷ hay chậm phát triển 4 .
Hội chứng Rett chủ yếu ảnh hưởng đến nữ giới. Theo thống kê, tỉ lệ mắc bệnh là 1:10,000 đến 1:15,000 không phân biệt chủng tộc, sắc tộc, vị trí địa lý. Hiếm ca bệnh Rett được ghi nhận ở nam giới, do những tổn hại quá lớn gây ra bởi những bất thường của yếu tố di truyền trước khi trẻ chào đời hoặc chết rất non 3 , 5 .
Cho đến nay, nguyên nhân di truyền gây ra Hội chứng Rett có liên quan nhất và được các nhà di truyền học đặc biệt quan tâm nghiên cứu là các biến thể de novo trên gen Methyl-CpG-binding Protein 2 hay gọi tắt là MECP2 6 , 7 . Biến thể ở gen này là nguyên nhân gây hội chứng Rett với tỉ lệ rất cao khoảng trên 92% trong hội chứng Rett điển hình và 50-70% ở hội chứng Rett không điển hình 8 , 9 . Một số bệnh nhân Rett không tìm thấy biến thể trên gen MECP2 có biến thể rải rác trên các gen khác như CDKL5 , FOXG1 10 , 11 . Các nghiên cứu trên thế giới vẫn đang tiếp tục giải thích và làm rõ các bất thường gen gây ra hội chứng Rett. Xét nghiệm MECP2 được xem như một tiêu chuẩn cận lâm sàng tin cậy hỗ trợ trong chẩn đoán hội chứng Rett trên thế giới.
MECP2 là một protein có vai trò quan trọng trong tất cả các tế bào của người, nhất là các tế bào thần kinh. Sự thiếu hụt hay dư thừa của MECP2 đều dẫn đến các rối loạn bệnh lý. Thiếu hụt MECP2 được ghi nhận trong các hội chứng Rett, hội chứng Angleman, tự kỷ, thiểu năng, bệnh não sơ sinh, hội chứng Down, hội chứng Prader Willi, rối loạn tăng động giảm chú ý 4 . … Sự nhân lên của gen MECP2 được phát hiện gây nên chứng thiểu năng, kém hoạt động, nhiễm trùng đường hô hấp, không phát triển khả năng giao tiếp bằng lời nói, động kinh và liệt cứng ở nam giới 5 . Bởi vì, MECP2 được biết đến như protein đa chức năng, ảnh hưởng đến quá trình điều hòa biểu hiện gen và quá trình trao đổi chất 12 .
Gen MECP2 mã hóa cho protein MECP2 (Methyl CpG binding Protein 2) chuyên bám vào DNA đã được methyl hóa nhằm điều hòa biểu hiện của gen. Quá trình methyl hoá cho phép tắt mở phiên mã các gen đó. Vì vậy, những biến đổi trên gen MECP2 sẽ gây ảnh hưởng lên chức năng của các gen khác 13 . Đặc biệt, gen MECP2 liên kết với nhiễm sắc thể (NST) giới tính X ở vị trí q28 do đó có thể làm bất hoạt NST này. Gen trải dài trên 76 kb của bộ gen và có 4 exon. Gen MECP2 mã hóa cho nhiều đồng phân protein, quan trọng nhất là 2 đồng phân MECP2 e1 và MECP2 e2 14 . Nghiên cứu ảnh hưởng của yếu tố di truyền đầu tiên tiến hành trên đồng phân MECP2e2 . Tuy nhiên, các nghiên cứu gần đây tìm thấy đột biến trên exon 1 mã hoá cho đồng phân MECP2e1 cũng là nguyên nhân gây ra hội chứng Rett 15 , 16 , 17 .
Khoảng 1000 biến thể trên gen MECP2 đã được công bố cho tới nay 18 . Trong đó, có 8 vị trí hot-spot được tìm thấy trong hội chứng này. Hiện tại, ở nước ta nghiên cứu trên hội chứng Rett còn khá hạn chế, trong đó mới chỉ có 1 nghiên cứu phân tích biến thể gen MECP2 trên bệnh nhân Rett. Tuy nhiên, nghiên cứu này đã bỏ qua việc phân tích đột biến trên exon 1 trong khi nhiều nghiên cứu gần đây trên thế giới cho thấy rằng đột biến trên exon 1 được ghi nhận trong nhiều bệnh nhân mắc hội chứng Rett. Do vậy, chúng tôi tiến hành nghiên cứu tối ưu hoá quy trình PCR- Sanger sequencing để giải toàn bộ 4 exon gen MECP2 . Từ đó, nghiên cứu này tạo tiền đề cho việc chẩn đoán sớm và tư vấn di truyền; đồng thời xây dựng dữ liệu dịch tể học phân tử về hội chứng Rett liên quan đến biến thể gen MECP2 trên dân số Việt Nam.
ĐỐI TƯỢNG - PHƯƠNG PHÁP NGHIÊN CỨU
Thiết kế nghiên cứu
Nguyên cứu cắt ngang mô tả.
Đối tượng nghiên cứu
Nghiên cứu bao gồm xây dựng và tối ưu quy trình trên mẫu đối chứng người bình thường. Sau đó áp dụng quy trình đã tối ưu lên 4 bệnh nhi mắc hội chứng Rett. Tất cả bệnh nhi được chẩn đoán bởi bác sĩ chuyên khoa thần kinh theo tiêu chuẩn chẩn đoán sửa đổi và bổ sung năm 2010 2 (phụ lục 1). Tất cả bệnh nhi là nữ và được điều trị tại bệnh viện Nhi đồng 2 Tp. Hồ Chí Minh từ tháng 1/2019 đến tháng 12/2019.
Đề tài được thông qua bởi hội đồng Y Đức Bệnh viện Nhi Đồng II, mã số CS/N2/18/01HT.
Phương pháp nghiên cứu
Tách chiết DNA từ máu ngoại vi
200µl máu tĩnh mạch đựng trong các tube có chứa EDTA chống đông được tách và trữ đúng quy cách. Quy trình tách chiết được thực hiện bằng bộ kit QIAamp DNA Blood Mini Kit của QIAGEN theo hướng dẫn của nhà sản xuất.
Khảo sát và tối ưu hóa phản ứng PCR
Sáu cặp mồi được thiết kế bằng phần mềm CLC main workbench (CLC bio) kết hợp với phần mềm thiết kế mồi Primer3 Plus dựa trên trình tự NG_0071072 và được kiểm tra độ đặc hiệu bằng phần mềm PrimerBlast. Sử dụng phần mềm Oligo analyzer (Integrated DNA Technologies, Inc.) kiểm tra các thông số hoạt động và độ nhạy của mồi.
Phản ứng PCR được thực hiện bởi 2 bộ kit là QIAGEN Multiplex PCR Kit cho exon 1 giàu GC và Q5® High-Fidelity 2X Master Mix, bao gồm các thành phần dNTP, Mg 2+ , DNA polymerase và các hóa chất đệm, …
Chúng tôi tiến hành tối ưu hoá nhiệt độ bắt cặp của mồi, thành phần phản ứng cũng như dung dịch đệm để đạt hiệu quả tốt nhất của phản ứng.
Giải trình tự và phân tích kết quả
Mẫu sau được khuếch đại sẽ đem giải trình tự trên máy 3130 AB Biosystems. Hoá chất sử dụng BigDye™ Terminator v3.1 Cycle Sequencing (hãng Thermo FirsherScientific).
Phân tích kết quả giải trình tự bằng phần mềm CLC Mainworkbench (CLC bio) với trình tự tham khảo cDNA đầy đủ của gen MECP2 được lấy từ GenBank (mã số cho biến thể MECP2e2 : NM_004992; mã số cho biến thể MECP2e1 : NM_001110792).
Sử dụng tiêu chuẩn phân loại và đánh giá khả năng gây bệnh của biến thể theo Hiệp Hội Di truyền Y học và Hệ Gen Hoa Kỳ (American College of Medical Genetics and Genomics (ACMG) 19 .
KẾT QUẢ VÀ THẢO LUẬN
Thu thập mẫu bệnh phẩm và tách chiết DNA
Nghiên cứu này bao gồm 4 bệnh nhi nữ mắc hội chứng Rett được chuẩn đoán và điều trị tại bệnh viện Nhi Đồng 2 Tp HCM. Kết quả tách chiết DNA cho thấy tất cả các mẫu tách chiết đều đạt nồng độ từ 40 – 60 µg/ml và độ tinh sạch cao (OD260/280 = 1,8–1,9).
Kết quả tối ưu phản ứng PCR
Chúng tôi khảo sát nhiệt độ bắt cặp (55ºC - 70ºC) dựa vào nhiệt độ nóng chảy của mồi và các thành phần của phản ứng. Kết quả thu nhận nhiệt độ tối ưu của các cặp mồi tương ứng với exon 2, 3, 4C là 63ºC, exon 4A là 62ºC, exon 4B là 67ºC, exon 1 là 59ºC. Thành phần của phản ứng đạt nồng độ cuối khuếch đại exon 1, 2, 3, 4A, 4B, 4C bao gồm mồi ngược và mồi xuôi: 0,5mM; MasterMix: 1X; DNA: 1,5ng/µL; nước denuclease đến thể tích cần thiết của phản ứng. Trong đó, exon 1 giàu GC, để thực hiện phản ứng PCR chúng tôi sử dụng thêm dung dịch đệm Q-solution của QIAGEN (chứa DMSO). Kết quả khuếch đại gen sau PCR được kiểm tra bằng điện di ( Figure 1 ), cho thấy cả 6 cặp mồi đều khuếch đại đặc hiệu và sản phẩm PCR đủ điều kiện để sử dụng cho quy trình giải trình tự. Danh sách 6 cặp mồi đã tối ưu như Table 1 .
Figure 1 . Kết quả điện di 6 sản phẩm PCR của gen MECP2. L: thang 1kb; 1: exon 1; 2: exon 2; 3: exon 3; 4,5,6: exon 4

Kết quả giải trình tự và phân tích gene
Gen MECP2 được giải trình tự toàn bộ 4 exon bằng kỹ thuật Sanger tự động cho kết quả tín hiệu tốt và ổn định được ghi nhận trên mẫu đối chứng. Sau đó chúng tôi áp dụng quy trình lên 04 mẫu bệnh nhi tìm thấy các biến thể trên gen MECP2 ( Figure 2 , Figure 3 , Figure 4 , Figure 5 ).
Figure 2 . Biến thể c.880C>T ở bệnh nhi số 1 thay đổi protein Arginine thành codon dừng phiên mã.

Figure 3 . Biến thể c.85A>T ở bệnh nhi số 2 thay đổi protein Lysine thành codon dừng phiên mã.
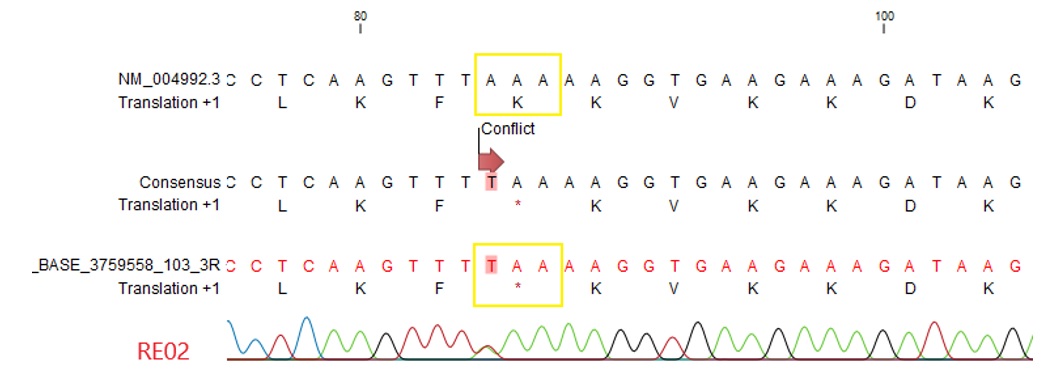

Chúng tôi tìm thấy 2 biến thể dừng phiên mã, bao gồm: c.880C>T (hình 2) (NM_004992) và c.85A>T (hình 3) (NM_004992) ở bệnh nhi số 1 và số 2. Biến thể c.880C>T trên exon 4 làm thay đổi trình tự acid amin thứ 294 trong chuỗi polypeptide từ Arginine thành codon dừng phiên mã (p.R294X). Biến thể c.85A>T, làm thay đổi amino acid tại vị trí 29 amino acid Lysine thành codon dừng phiên mã (p.K29X).
Áp dụng tiêu chuẩn ACMG để phân loại biến thể, kết quả cho thấy 2 biến thể vùng exon đều là biến thể gây bệnh. Đột biến c.880C>T là đột biến có tần số cao chiếm gần 5% trong tổng số đột biến tìm thấy trên gen MECP2 đã được công bố gây hội chứng Rett 18 . Cơ sở dữ liệu ClinVar ghi nhận biến thể với mã rs61751362, và thống kê cho thấy hầu hết các nghiên cứu đều phân tích đây là biến thể gây bệnh. Riêng đột biến dừng phiên mã sớm c.85A>T là đột biến gây bệnh mới nằm trên domain N-term và chưa từng được công bố.
Ngoài ra, chúng tôi tìm thấy 4 biến thể không gây bệnh (SNV) trên 3 bệnh nhi nữ bao gồm: c.602C>T trên bệnh nhi số 2, biến thể vùng intron: c.377+13A>C (hình 4), c.377+22C>G (hình 4), c.378-74C>T (hình 5) trên bệnh nhân 3 và bệnh nhân 4.
Các vị trí SNV c.602C>T, c.377+22C>G, c.378-74C>T đã được ghi nhận trên thế giới 18 . Riêng biến thể c.377+13A>C là biến thể lành tính lần đầu tiên được ghi nhận trong nghiên cứu này. Khảo sát đặc tính di truyền cho thấy tất cả các biến thể này được di truyền từ người bố. Sử dụng công cụ InterVar (phân loại biến thể theo tiêu chuẩn ACMG) để dự đoán ảnh hưởng của các biến thể, kết quả cho thấy đây là các biến thể không gây bệnh. Trong đó, các biến thể xuất hiện trên chủng người Việt với tần số như sau c.602 C>T (rs61748381 20 là 0.03; c.377+22 C>G (rs2075597 20 là 0,128; c.378-74C>T (rs2071569 20 . Các biến thể này được ghi nhận trước đó cho thấy chúng xuất hiện trong hội chứng Rett, tự kỷ, và các hội chứng rối loạn nhận thức không thường xuyên.
Biến thể trên gen MECP2 đã được báo cáo là nguyên nhân gây ra khoảng hơn 92% các trường hợp mắc hội chứng Rett điển hình và khoảng 50 -70% hội chứng Rett không điển hình 18 . Hiện nay, khoảng 1000 biến thể MECP2 đã được phát hiện và phần lớn là đột biến điểm. Trong nghiên cứu này, chúng tôi đã ghi nhận hai biến thể p.R294X (cắt cụt gần 45% protein), p.K29R (cắt cụt gần 95% protein) làm mất chức năng của protein MECP2 . Về mặt lâm sàng, hai bệnh nhi Rett mang hai biến thể này có những đặc điểm kiểu hình Rett điển hình, xét nghệm gen khi đã ở giai đoạn muộn khi bệnh nhân số 1 mười tuổi và bệnh nhân số 2 sáu tuổi. Hai bệnh nhân này mất ngôn ngữ và mất khả năng giao tiếp; mất khả năng điều khiển tay, không thực hiện được các hoạt động tay phối hợp. Có các cử động tay bất thường không chủ đích: đưa tay vào miệng, thực hiện thao tác xoắn vặn bàn tay ngón tay; khó khăn và không thể đi đứng bình thường được, hay té ngã do rối loạn trương cơ; bất thường kiểu thở với đặc điểm thở mạnh và chảy nhiều nước bọt; phát triển thể chất bất thường đặc biệt là gù lưng và một số biểu hiện không đặc hiệu khác như gầy ốm, hay la hét, dễ kích động…
Gần đây, nghiên cứu về biến thể gen MECP2 trên hội chứng Rett ở nước ta chỉ còn rất hạn chế và chỉ mới tập trung vào các exon 2, 3 và 4 21 . Do vậy, nghiên cứu này là bước đầu để phân tích thay đổi trên toàn bộ 4 exon của gen MECP2 ở các bệnh nhân mắc hội chứng Rett; từ đó, thiết lập được quy trình xét nghiệm gen MECP2 nhanh, tiết kiệm và đáng tin cậy, góp phần phát hiện và tầm soát biến thể gen liên quan đến hội chứng Rett, giảm bớt gánh nặng cho các gia đình có bệnh nhân mắc bệnh. Nghiên cứu được thực hiện trên số mẫu khá hạn chế, do vậy đây chỉ là nghiên cứu khởi đầu cho bước tiếp theo thực hiện nghiên cứu trên số lượng mẫu lớn để ứng dụng triển khai trên lâm sàng. Tuy vậy, trong nghiên cứu này, chúng tôi đã bổ sung được 1 biến thể gây bệnh (c.85A>T) và 1 SNV (c.377+13A>C) mới vào ngân hàng dữ liệu biến thể gen MECP2 thế giới.
KẾT LUẬN
Chúng tôi đã xây dựng thành công và hoàn thiện quy trình phát hiện đột biến điểm trên gen MECP2 bằng kỹ thuật PCR-sequencing trên các bệnh nhi mắc hội chứng Rett. Nghiên cứu này là cơ sở để triển khai khảo sát biến thể gen MECP2 trên bệnh nhân hội chứng Rett.
Nghiên cứu này được tài trợ bởi Đại học Quốc gia TP.HCM (Mã số đề tài C2018-44-02).
DANH MỤC CÁC TỪ VIẾT TẮT
MECP2 : Methyl CpG binding Protein 2
PCR : Polymerase Chain Reaction
ACMG : American College of Medical Genetics and Genomics
SNV: Single Nucleotide Variant
c.880C>T : Biến thể tại vị trí 880 trên vùng mã hoá gen làm thay đổi nucleotide C thành T so với gen tham chiếu
c.85A>T : Biến thể tại vị trí 85 trên vùng mã hoá gen làm thay đổi nucleotide C thành T so với gen tham chiếu
c.602C>T : Biến thể tại vị trí 602 trên vùng mã hoá gen làm thay đổi nucleotide C thành T so với gen tham chiếu
c.377+13A>C : Biến thể tại vùng intron trên cách vùng mã hoá 13 nucleotide tại vị trí 377 làm thay đổi nucleotide A thành C so với gen tham chiếu
c.377+22C>G : Biến thể tại vùng intron trên cách vùng mã hoá 22 nucleotide tại vị trí 377 làm thay đổi nucleotide A thành T so với gen tham chiếu
c.378-74C>T : Biến thể tại vùng intron trên cách vùng mã hoá 74 nucleotide tại vị trí 378 làm thay đổi nucleotide A thành T so với gen tham chiếu
p.R294X : Acid amin Arginine tại vị trí 294 bị thay đổi thành codon dừng phiên mã so với protein tham chiếu
p.K29R : Acid amin Lysin tại vị trí 29 bị thay đổi thành codon dừng phiên mã so với protein tham chiếu
XUNG ĐỘT LỢI ÍCH
Nhóm tác giả cam kết rằng không có xung đột lợi ích khi thực hiện nghiên cứu này.
ĐÓNG GÓP CỦA TÁC GIẢ
Tất cả tác giả đã đóng góp vào việc thiết kế nghiên cứu, giải thích kết quả nghiên cứu, viết và chỉnh sửa bản thảo của bài báo, xem xét cẩn thận đồng ý nộp bản thảo hoàn chỉnh này.
ĐẠO ĐỨC TRONG NGHIÊN CỨU Y SINH
Nghiên cứu thuộc đề tài C2018-44-02 ĐHQG TPCHM của KHOA Y ĐHQG TPHCM đã được phê duyệt về các vấn đề đạo đức trong nghiên cứu y sinh học, chấp thuận số CS/N2/18/01HT của Hội đồng khoa học/Y đức Bệnh Viện Nhi Đồng II.
Phụ lục 1
Tiêu chuẩn chẩn đoán bệnh nhân mắc hội chứng Rett thu mẫu cho nghiên cứu này 2 .
Chỉ tiêu đánh giá
Tiêu chuẩn chọn bệnh nhân:
Bệnh nhân được chẩn đoán mắc hội chứng Rett cả điển hình hoặc Rett không điển hình.
A. Bệnh nhân được chẩn đoán mắc hội chứng Rett điển hình khi:
(1) Bệnh nhân tồn tại một giai đoạn phục hồi (hồi quy)
(2) Có tất cả các tiêu chuẩn chính (bảng bên dưới) và không bao gồm các tiêu chuẩn loại trừ (bảng bên dưới).
(3) Có các tiêu chuẩn phụ trợ đi kèm, điều này là không bắt buộc tuy nhiên trẻ thường sẽ có hầu hết các đặc điểm trong tiêu chuẩn phụ trợ.
B. Bệnh nhân được chẩn đoán mắc hội chứng Rett không điển hình khi:
(1) Bệnh nhân tồn tại một giai đoạn phục hồi (hồi quy)
(2) Ít nhất 2 trong số 4 tiêu chuẩn chính
(3) 5 trên 11 tiêu chuẩn phụ trợ
Ngoài ra tiêu chuẩn: giảm kích thước vòng đầu của trẻ chưa được xem xét trong bảng tiêu chuẩn này. Ở đây, chúng tôi tạm đánh giá tiêu chuẩn này là 1 tiêu chuẩn phụ trợ.
References
- Rett A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wiener Medizinische Wochenschrift. 1966;116(37):723-726. Google Scholar
- Jeffrey LN, Walter EK, Daniel GG, John C, Angus JC, Alan KP, et al. RettSearch 'Rett Syndrome: Revised Diagnostic Criteria and Nomenclature'. Ann Neurol. 2010;68:944-950. PubMed Google Scholar
- Rett A. Rett Syndrome. History and General Overview. Am J Med Genet Suppl. 1986;1:21-25. PubMed Google Scholar
- Matthew JL, Adrian B, Rett Syndrome: A Complex Disorder with Simple Roots. Nat Rev Genet. 2015;16:261-275. PubMed Google Scholar
- Kankirawatana P, Leonard H, Ellaway C, Scurlock J, Percy AK, et al. Early Progressive Encephalopathy in Boys and MECP2 Mutations. Neurology. 2006;67:164-166. PubMed Google Scholar
- Amir RE, Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett Syndrome Is Caused by Mutations in X-Linked MECP2, Encoding Methyl-Cpg-Binding Protein 2. Nat Genet. 1999;23:185-188. PubMed Google Scholar
- Webb T, Latif F. Rett Syndrome and the MECP2 Gene. J Med Genet. 2001;31:217-223. PubMed Google Scholar
- Percy AK, Lane JB, et al. Rett Syndrome: Clinical and Molecular Update. Curr Opin Pediatr. 2004;16:670-677. PubMed Google Scholar
- Percy AK, Lane JB, Childers J, Skinner S, Annese F, MacLeod P. Rett Syndrome: North American Database. J Child Neurol. 2007;22:1338-1341. PubMed Google Scholar
- Ariani F, Hayek G, Rondinella D, Artuso R, Renieri A, et al. Foxg1 Is Responsible for the Congenital Variant of Rett Syndrome. Am J Hum Genet. 2008;83:89-93. PubMed Google Scholar
- Bahi-Buisson N, Nectoux J, Rosas-Vargas H, Milh M, Bienvenu T, et al. Key Clinical Features to Identify Girls with Cdkl5 Mutations. Brain. 2008;131:2647-2661. PubMed Google Scholar
- Friez MJ, Jones JR, Clarkson K, Lubs H, Stevenson RE, et al. Recurrent Infections, Hypotonia, and Mental Retardation Caused by Duplication of MECP2 and Adjacent Region in Xq28. Pediatrics. 2006;118:e1687-e1695. PubMed Google Scholar
- Nagarajan RB, Hogart AR, Gwye Y, Martin MR, LaSalle JM. Reduced MECP2 Expression Is Frequent in Autism Frontal Cortex and Correlates with Aberrant MECP2 Promoter Methylation. Epigenetics. 2006;1:e1-e11. PubMed Google Scholar
- Reichwald K, Thiesen J, Wiehe T, Weitzel A, Platzer M, et al. Comparative sequence analysis of the MECP2-locus in human and mouse reveals new transcribed regions. Mammalian Genome. 2000;11:182-190. PubMed Google Scholar
- Aline Q, SalihaY, Hervé F, Thierry B, Anne M, Christophe P, et al. Deleterious mutations in exon 1 of MECP2 in Rett syndrome. European Journal of Medical Genetics. 2006;49(4):313-322. PubMed Google Scholar
- Carol JS, Berge EM, Eva WCC, Weiwei Z, John BV. Novel exon 1 mutations in MECP2 implicate isoform MeCP2_e1 in classical Rett syndrome. Am J Med Genet. 2009;149A:1019-1023. PubMed Google Scholar
- Bartholdi D, Klein A, Weissert M, Koenig N, Mátyás G, et al. Clinical profiles of four patients with Rett syndrome carrying a novel exon 1 mutation or genomic rearrangement in the MECP2 gene. Clin Genet. 2006;69(4):319-326. PubMed Google Scholar
- Krishnaraj R, Ho G, Christodoulou J. RettBASE: Rett syndrome database update. Hum Mutat. [RettBASE: RettSyndrome.org Variation Database mecp2.chw.edu.au]. 2017;:. PubMed Google Scholar
- Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. PubMed Google Scholar
- . . ;:. Google Scholar
- Huong LTT, Trinh DTD, Chinh VD, Ha LTT, Hoa BTP, and Liem NT. Spectrum of MECP2 mutations in Vietnamese patients with RETT syndrome. BMC Med Genet. 2018;19:137. PubMed Google Scholar




